Research Overview
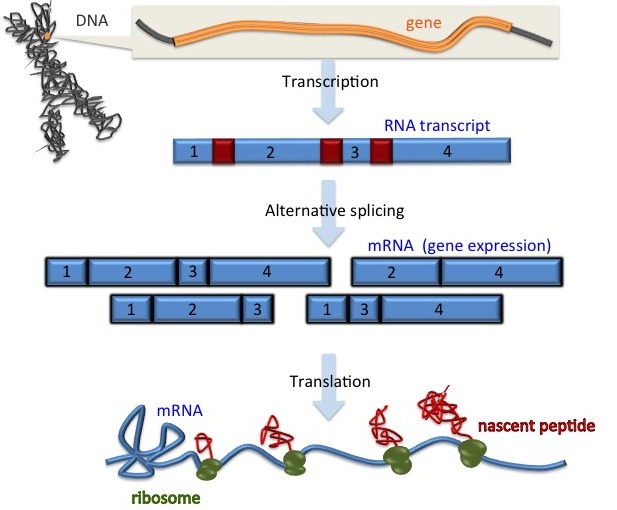
My research centers around statistical learning motivated by computational biology and biomedical research. From a bioinformatics point of view, I have been studying gene regulation, including both transcription and translation, heterochromatin dynamics, and automated feature extraction for time series in electrocardiology and volumetric 3D images for cell deformation. From a machine learning point of view, I am interested in high-dimensional and small sample size problems, optimization, structured variable selection, density estimation, functional data analysis, neural network. The top figure shows gene regulation, and a brief overview of my research topics are categorized as follows, each one focuses on different ascepts of gene regulation highlighted in grey. However, one should note that many of these research topics are multidisciplinary, and are made possible by the strength of both statistical signal processing/computation and biomedical research. The categories described below are artificial for convenience reasons, and it is sometimes difficult to assign some of the research results into a single category.
Predictive Health Study
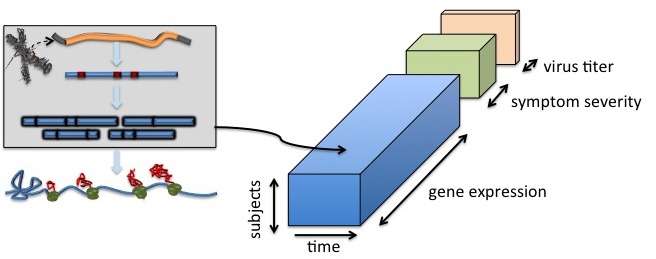 Consider the problem of predicting symptom severity based on gene expression, in which the dimension of gene expression is much larger than the number of samples. Dimension reduction methods that incorporate biomarker screening and prediction would be useful in these problems. Partial least squares (PLS) regression is a supervised dimension reduction method, which incorporates prediction into dimension reduction. It does not involve matrix inversion nor diagonalization. Hence, it has been successfully applied to problems with large predictor dimension. However, as the number of predictor variables increases, PLS can suffer from over-fitting, i.e., the prediction performance degrades, and the parameters become difficult to interpret. A global variable selection approach was proposed, which penalized the total number of variables across all PLS components. Results showed that the proposed formulation successfully reduced model complexity by selecting many fewer predictor variables, while achieving good prediction ability.
Consider the problem of predicting symptom severity based on gene expression, in which the dimension of gene expression is much larger than the number of samples. Dimension reduction methods that incorporate biomarker screening and prediction would be useful in these problems. Partial least squares (PLS) regression is a supervised dimension reduction method, which incorporates prediction into dimension reduction. It does not involve matrix inversion nor diagonalization. Hence, it has been successfully applied to problems with large predictor dimension. However, as the number of predictor variables increases, PLS can suffer from over-fitting, i.e., the prediction performance degrades, and the parameters become difficult to interpret. A global variable selection approach was proposed, which penalized the total number of variables across all PLS components. Results showed that the proposed formulation successfully reduced model complexity by selecting many fewer predictor variables, while achieving good prediction ability.
- T.-Y. Liu, L. Trinchera, A. Tenenhaus, D. Wei, and A. O. Hero. Jointly Sparse Global SIMPLS Regression, submitted.[html]
- M. T. McClain, B. P. Nicholson, L. P. Park, T.-Y. Liu, A. O. Hero III, E. Tsalik, A. K. Zaas, T. Veldman, L. L. Hudson, R. Lambkin-Williams, A. Gilbert, G. S. Ginsburg and C. W. Woods. A Genomic Signature of Influenza Infection Shows Potential for Presymptomatic Detection, Guiding Early Therapy, and Monitoring Clinical Responses, Open Forum Infectious Diseases, Vol. 3, No. 1, doi:10.1093/ofid/ofw007, 2016. [html]
- T.-Y. Liu, L. Trinchera, A. Tenenhaus, D. Wei, and A. O. Hero. Globally Sparse PLS Regression. New Perspectives in Partial Least Squares and Related Methods, Springer Proceedings in Mathematics and Statistics, Vol. 56, pp. 117-127, 2013. [html]
Personalized Medicine
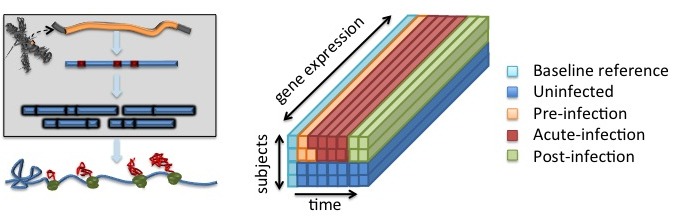 Consider the problem of designing a panel of complex biomarkers to predict a patient's health or disease state when one can pair his or her current test sample, called a target sample, with the patient's previously acquired healthy sample, called a reference sample. As contrasted to a population averaged reference this reference sample is individualized. I introduced a sparsity penalized multi-class classifier design to account for multi-block structure of the data, which arises naturally in serially sampled data or spatially diversified sampling experiments. The classifier was trained to minimize an objective function that captures the unified miss-classification probabilities of error over the classes in addition to the sparsity of the weights. Results showed that the disease prediction rate was improved and the method was able to control irrelevant patient variations.
Consider the problem of designing a panel of complex biomarkers to predict a patient's health or disease state when one can pair his or her current test sample, called a target sample, with the patient's previously acquired healthy sample, called a reference sample. As contrasted to a population averaged reference this reference sample is individualized. I introduced a sparsity penalized multi-class classifier design to account for multi-block structure of the data, which arises naturally in serially sampled data or spatially diversified sampling experiments. The classifier was trained to minimize an objective function that captures the unified miss-classification probabilities of error over the classes in addition to the sparsity of the weights. Results showed that the disease prediction rate was improved and the method was able to control irrelevant patient variations.
- T.-Y. Liu, T. Burke, L. P. Park, C. W. Woods, A. K. Zaas, G. S. Ginsburg and A. O. Hero. An individualized predictor of health and disease using paired reference and target samples, BMC bioinformatics, Vol. 17, No. 1, pp. 1-15, 2016. [html]
- T.-Y. Liu, A. Wiesel, and A. O. Hero. "A Sparse Multi-class Classifier for Biomarker Screening". Global Conference on Signal and Information Processing (GlobalSIP) Proceedings, IEEE, pp. 77-80, 2014.[html]
- T.-Y. Liu, A. O. Hero. A Structured Sparse Multi-class Classifier, to be submitted.
Translational Dynamics - Prediction of Ribosome Densities
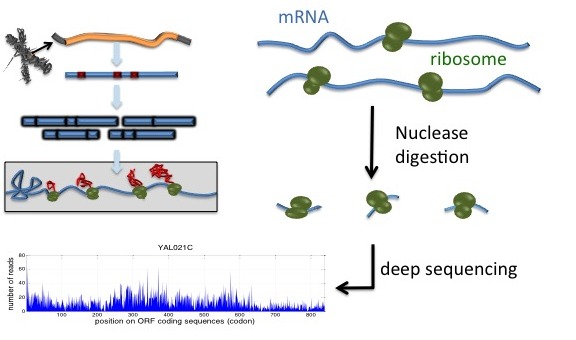 The ribosomes are not uniformly distributed along the transcripts. Understanding how transcript-specific distribution arises and to what extent it depends on the sequence contents is fundamental for unraveling the translation mechanism. Motivated by the observed profiles of ribosome footprints in the literature, which seem to distribute far from uniformly, and the different hypotheses explaining the underlying mechanism in the literature, here I focus on the prediction of marginal densities of ribosome footprints using solely the sequence context. This is an interesting machine learning problem, in which the predictors are categorical, and the response variables are continuous. The ability to predict the marginal densities based on the sequence contents alone has many potential applications in various areas, including isoform specific ribosome inference, design of transcripts with fast translation, etc.
The ribosomes are not uniformly distributed along the transcripts. Understanding how transcript-specific distribution arises and to what extent it depends on the sequence contents is fundamental for unraveling the translation mechanism. Motivated by the observed profiles of ribosome footprints in the literature, which seem to distribute far from uniformly, and the different hypotheses explaining the underlying mechanism in the literature, here I focus on the prediction of marginal densities of ribosome footprints using solely the sequence context. This is an interesting machine learning problem, in which the predictors are categorical, and the response variables are continuous. The ability to predict the marginal densities based on the sequence contents alone has many potential applications in various areas, including isoform specific ribosome inference, design of transcripts with fast translation, etc.
- T.-Y. Liu, Y. S. Song. Prediction of Ribosome Footprint Profile Shapes from Transcript Sequences, Bioinformatics,Vol. 32, No. 12, pp. i183-i191, doi:10.1093/bioinformatics/btw253, 2016. [html]
- T.-Y. Liu, Y. S. Song. "Prediction of Ribosome Footprint Distributions from Transcript Sequences via Multiresolution Analysis". Neural Information Processing Systems (NIPS) workshop on Machine Learning in Computational Biology (MLCB), 2015 (oral Presentation).
- T.-Y. Liu, Y. S. Song. "Can you predict the shape of Ribosome profiles? Marginal Probability Density Estimation of Ribosome Footprints". Cold Spring Harbor Laboratory (CSHL) Probabilistic Modeling in Genomics, 2015.
Integrative Longitudinal Analysis of Ribosome Occupancy and Protein Synthesis
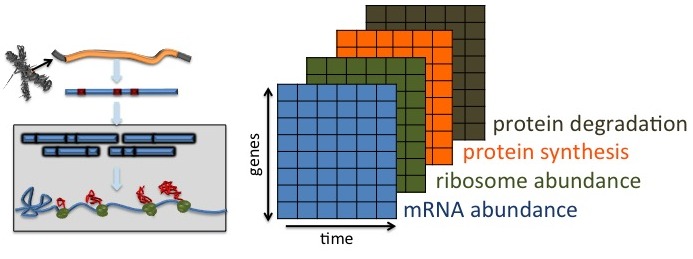 The regulation of gene expression is composed of transcription and translation. During translation, ribosomes traverse each codon of the mRNA transcripts to synthesize proteins according to the message encoded in the transcripts. Although much had been studied in the transcription level with the advances in microarray and deep sequencing, studies of the translational dynamics remained challenging until the development of ribosome profiling. Ribosome profiling provides a snapshot of the distribution of these ribosomes along transcripts and enables quantitative monitoring and analysis of the translational process. In this project, a collaboration with Dr. Arun Wiita's lab at UCSF, I developed functional data analysis methods that jointly analyze mRNA-seq, ribosome profiling, and pulse-chase isotopic labeling mass spectrometry-based proteomics. Our work offers a novel quantitative framework to understand translation using a combination of emerging technologies. Taking advantage of this model with concurrent biochemical and genetic experimentation may allow us to identify these factors that govern translational regulation in cancer and potentially eukaryotes more broadly, and shed light on targeted therapies.
The regulation of gene expression is composed of transcription and translation. During translation, ribosomes traverse each codon of the mRNA transcripts to synthesize proteins according to the message encoded in the transcripts. Although much had been studied in the transcription level with the advances in microarray and deep sequencing, studies of the translational dynamics remained challenging until the development of ribosome profiling. Ribosome profiling provides a snapshot of the distribution of these ribosomes along transcripts and enables quantitative monitoring and analysis of the translational process. In this project, a collaboration with Dr. Arun Wiita's lab at UCSF, I developed functional data analysis methods that jointly analyze mRNA-seq, ribosome profiling, and pulse-chase isotopic labeling mass spectrometry-based proteomics. Our work offers a novel quantitative framework to understand translation using a combination of emerging technologies. Taking advantage of this model with concurrent biochemical and genetic experimentation may allow us to identify these factors that govern translational regulation in cancer and potentially eukaryotes more broadly, and shed light on targeted therapies.
- T.-Y. Liu*, H. H. Huang*, D. Wheeler, Y. Xu, J. A. Wells, Y. S. Song, A. P. Wiita. Time-resolved proteomics extends ribosomal profiling-based measurements of protein synthesis dynamics, Cell Systems, Vol. 4, No. 6, pp. 636-644, 2017. [html]
- T.-Y. Liu, H. H. Huang, D. Wheeler, Y. S. Song, A. P. Wiita. "Direct measurement and modeling of protein synthesis and degradation dynamics during chemotherapeutic response in multiple myeloma". American Society for Mass Spectrometry (ASMS), 2016.
- T.-Y. Liu, H. H. Huang, D. Wheeler, J. A. Wells, Y. S. Song, A. P. Wiita. "Integrative longitudinal analysis of ribosome occupancy and protein synthesis during chemotherapeutic response reveals complex translational dynamics". American Society of Human Genetics Annual Meeting (ASHG), 2015. * authors with equal contributions
Automated Analysis of Heterochromatin Dynamics
 The genome and physiology of a cell can undergo complex changes among the many cells that make up a growing microbial colony. Genetic and physiological dynamics can be revealed by measuring reporter-gene expression, but rigorous quantitative analysis of colony-wide patterns has been under-explored. In this collaboration with Dr. Jasper Rine's lab at UCB, I developed a suite of automated image processing, visualization, and classification algorithms (Morphological Phenotype Extraction: MORPHE) that facilitated the analysis of heterochromatin dynamics in the context of colonial growth and that can be broadly adapted to many colony-based assays in Saccharomyces and other microbes. Using the features that were automatically extracted from fluorescence images, MORPHE revealed subtle but significant differences in the stability of heterochromatic repression, which were not apparent by visual inspection.
The genome and physiology of a cell can undergo complex changes among the many cells that make up a growing microbial colony. Genetic and physiological dynamics can be revealed by measuring reporter-gene expression, but rigorous quantitative analysis of colony-wide patterns has been under-explored. In this collaboration with Dr. Jasper Rine's lab at UCB, I developed a suite of automated image processing, visualization, and classification algorithms (Morphological Phenotype Extraction: MORPHE) that facilitated the analysis of heterochromatin dynamics in the context of colonial growth and that can be broadly adapted to many colony-based assays in Saccharomyces and other microbes. Using the features that were automatically extracted from fluorescence images, MORPHE revealed subtle but significant differences in the stability of heterochromatic repression, which were not apparent by visual inspection.
- T.-Y. Liu*, A. E. Dodson*, J. Terhorst, Y. S. Song and J. Rine. Riches of Phenotype Computationally Extracted from Microbial Colonies, Proceedings of the National Academy of Sciences, Vol. 113, No. 20, pp. E2822-E2831, doi:10.1073/pnas.1523295113, 2016. [html] * authors with equal contributions
Automated Image Segmentation and Feature Extraction with Applications to Cell Deformation, Heterochromatin Dynamics
 Modern developments in light microscopy have allowed the observation of cell deformation with remarkable spatiotemporal resolution and reproducibility. Due to the considerable complexity of cell deformation and migration, visual analysis of such processes is not only limited by user bias, but also fails to apprehend large-scale, population-wise patterns that may otherwise appear random or disorganised. Systematic quantitative analysis and understanding of such phenomena is therefore becoming a major interest for the signal processing and computer vision communities. A combination of shape description, i.e., spherical harmonics analysis, and machine-learning techniques was proposed to analyze amoeboid cell spatiotemporal deformation, recorded as time-lapse sequences of volumetric 3D images.
Modern developments in light microscopy have allowed the observation of cell deformation with remarkable spatiotemporal resolution and reproducibility. Due to the considerable complexity of cell deformation and migration, visual analysis of such processes is not only limited by user bias, but also fails to apprehend large-scale, population-wise patterns that may otherwise appear random or disorganised. Systematic quantitative analysis and understanding of such phenomena is therefore becoming a major interest for the signal processing and computer vision communities. A combination of shape description, i.e., spherical harmonics analysis, and machine-learning techniques was proposed to analyze amoeboid cell spatiotemporal deformation, recorded as time-lapse sequences of volumetric 3D images.
- A. Dufour, T.-Y. Liu, C. Ducroz, R. Tournemenne, B. Cummings, R. Thibeaux, N. Guillen, A. O. Hero III, and J.-C. Olivo-Marin. Signal Processing Challenges in Quantitative 3-D Cell Morphology. IEEE Signal Processing Magazine, pp. 30-40, 2014. [html]
- T.-Y. Liu, M. Perlman, A. O. Hero, M. Roth, I. Rajapakse. Chromosome conformation during suspended animation and reversible stopping of biological time, in preparation.
Automated Analysis of Electrocardiogram to Identify the Origin of Arrythmia
Ventricular tachycardia (VT) is a potentially life-threatening arrhythmia that can lead to ventricular fibrillation and sudden death. Detecting and localizing VT are therefore important in the area of electrocardiology. The data consists of high dimensional time series with high variability. Developed algorithms that used single lead electrograms as a surrogate for 12 lead electrocardiograms and automated classification or prediction of the origin of VT based on electrocardiograms can result in a reduction of the time duration of the pace-mapping procedure, which usually takes more than 6 hours.- M. Yokokawa*, T.-Y. Liu*, K. Yoshida, C. Scott, A. O. Hero, E. Good, F. Morady, and F. Bogun. Automated analysis of the 12-lead electrocardiogram to identify the exit site of postinfarction ventricular tachycardia. Heart Rhythm, Vol. 9, No. 3, pp. 330-334, 2012. [html]
- K. Yoshida*, T.-Y. Liu*, C. Scott, A. Hero, M. Yokokawa, S. Gupta, E. Good, F. Morady, F. Bogun. The Value of Defibrillator Electrograms for Recognition of Clinical Ventricular Tachycardias and for Pace-Mapping Of Post-Infarction Ventricular Tachycardia. Journal of the American College of Cardiology, Vol. 56, No. 12, pp. 969-979, 2010. [html]
- T. S. Baman, D. C. Lange, K. J. Ilg, S. K. Gupta, T.-Y. Liu, C. Alguire, W. Armstrong, E. Good, A. Chugh, K. Jongnarangsin, F. Pelosi Jr., T. Crawford, M. Ebinger, H. Oral, F. Morady, F. Bogun, Relationship between burden of premature ventricular complexes and left ventricular function. Heart Rhythm Vol. 7, No. 7, pp. 865-869, 2010. [html] * authors with equal contributions